
神经退行性疾病的典型特征是神经系统特定(亚)细胞群的迟发性进行性损伤,这些细胞对活动、协调、力量、感觉和认知至关重要。随着单细胞组学技术的出现,解决这种选择性细胞脆弱性已经成为可能,单细胞组学技术现在代表了以前所未有的分辨率描绘复杂组织(包括人类死后大脑)异质性的最先进方法。在这篇综述中,我们简要概述了单细胞RNA测序的实验工作流程,并总结了最近在最常见的神经退行性疾病中获得的知识:帕金森病,阿尔茨海默病,亨廷顿病和多发性硬化症。我们还讨论了应用单细胞方法诊断和治疗神经退行性疾病的可能性,以及局限性。虽然我们目前正处于深入探索受影响细胞的转录组变化的阶段,但一旦我们更好地了解受影响的途径,进一步的技术发展有望操纵受影响的途径。
神经退行性疾病(ND)是由中枢神经系统细胞(如神经元)的进行性损失引起的,这一过程被称为神经变性[1]。它们影响着全世界数百万人,而且随着年龄的增长,特别是随着预期寿命的增加,患ND的可能性急剧上升。最常见的ND包括阿尔茨海默病(AD)、帕金森病(PD)、亨廷顿病(HD)、多发性硬化症(MS)和肌萎缩侧索硬化症(ALS)[2,3,4,5,6]。虽然在某些情况下,ND的病因可以通过众所周知的遗传改变来解释,但其他情况是特发性的,甚至是由环境因素引起的[7,8]。鉴于这些疾病大多表现出较晚的发病年龄(AAO)和进行性病程,了解病理改变之前的细胞机制一直是神经科学的长期目标,最终目的是纠正和/或预防它们[9]。
最初的神经解剖学和分子研究确定了受这些疾病影响的大脑区域以及致病事件:阿尔茨海默病主要受影响的是内吸皮层,淀粉样蛋白和tau蛋白沉积[2],帕金森病主要受影响的是黑质多巴胺能神经元的进行性丢失[3],而亨廷顿蛋白基因(HTT) CAG重复扩增引起的纹状体是HD的受影响组织[4](图1)。然而,这些大脑区域都由多种细胞类型组成,通常不止一个区域受到不同程度的影响。传统的方法,如免疫染色和组织学,往往缺乏必要的通量和分辨率,无法获得与ND相关的所有细胞类型和基因的公正的全局视图[10,11]。最近引入的单细胞基因组学方法为量化任何组织、器官或整个生物体中的细胞组成变化和基因表达变化提供了强大的工具(https://www.humancellatlas.org/)[12,13,14,15]。
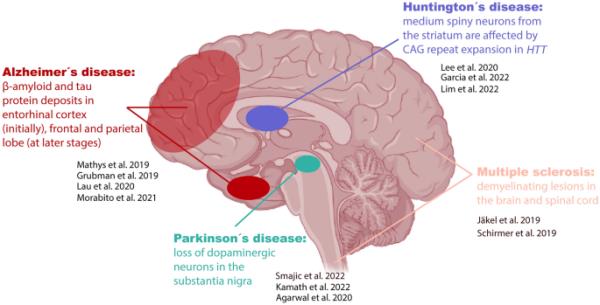
最常见的神经退行性疾病中受影响的大脑区域,以及关于sc/snRNA-seq的重要出版物
在这篇综述文章中,我们总结了利用单细胞基因组学方法了解人类死后脑组织中最常见的神经退行性疾病、诱导多能干细胞(iPSCs)和动物模型的最新进展,重点是帕金森病。
最近,单细胞和单核RNA测序技术的进步使得高分辨率的新鲜和冷冻组织分析成为可能,从而能够同时评估数千个细胞的转录组[16]。传统的批量RNA测序只能为测序的细胞群体提供平均的基因表达信号,与之相反,单细胞测序可以区分每个细胞的基因表达谱。通过保留细胞异质性,这种方法甚至可以检测到小的、疾病特异性的细胞亚群[17]。一般来说,工作流程从组织采集开始(图2a),然后确定是否将分离单个细胞或细胞核:细胞从新鲜组织中分离,而细胞核从冷冻组织(例如,大脑)中分离。组织采集是工作流程中的关键步骤,因为它决定了RNA的质量——就人脑而言,尸检间隔应尽可能短,组织解剖应在低温下进行,样品应快速冷冻,以避免RNA降解[15]。接下来,从组织中分离出单个细胞或细胞核(图2b),并将其包裹在孔或条形码液滴中(图2c)。在每个被封装的实体中,发生逆转录,随后是cDNA扩增(图2d)和下一代测序(图2e)。然后将Reads与参考基因组/转录组/表观基因组进行比对并进行量化[17]。定量数据被组织到特征条形码矩阵中,该矩阵在归一化后允许在细胞之间进行数据比较(图2f)。然后使用诸如主成分分析之类的方法对特征(通常有数千个特征)进行浓缩,从而降低数据维数。这些浓缩的数据然后被聚类,这是指具有相似特征的细胞分组,例如,根据它们的基因表达谱。聚类结果通常通过进一步降低维数来可视化,使用均匀流形逼近和投影(UMAP)或t分布随机邻居嵌入(t-SNE)等方法;图2 g)。这两种方法都将结果可视化为散点图,其中每个点代表一个细胞,并且这些细胞在接近时将具有相似的基因表达谱。通常,每个集群代表一种细胞类型,并且可以进一步细分为细胞亚群。细胞(亚)簇的注释是基于文献中已知的给定簇中高表达的基因进行的(例如,少突胶质细胞以高表达MOPB为特征;星形胶质细胞受AQP4影响;多巴胺能神经元通过TH等)[15,18,19,20]。Heumos等人最近发表了一篇关于跨模式单细胞分析最佳实践的详细综述[21]。
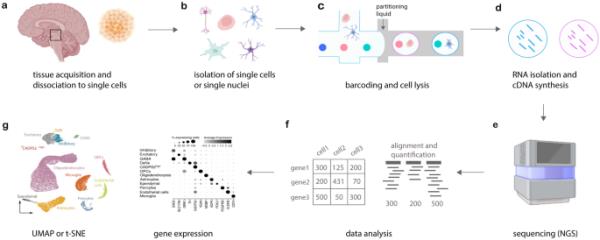
scRNA-seq的工作流程包括组织采集和b单细胞/细胞核的分离,c条形码和细胞裂解,d RNA分离和逆转录,e测序,f数据分析,g结果可视化
帕金森病(PD)是一种进行性神经退行性疾病,其特征是运动性(运动迟缓合并静止震颤和/或僵硬)和非运动性症状(认知能力下降、抑郁、疼痛)的结合[3]。只有5%的患者可以找到疾病的遗传原因,其余大多数患者没有明确的遗传原因,因此被归类为特发性PD[22]。该疾病的神经病理学特征是存在路易小体和路易神经突,即含有蛋白α-突触核蛋白的神经元包涵体。路易氏病理包括囊泡结构、线粒体等畸形细胞器和高脂质含量[23]。这些结构的功能障碍和神经炎症导致位于黑质的多巴胺能神经元(DaNs)死亡[3]。尽管大多数PD研究都集中在DaNs上,但其他类型的细胞如小胶质细胞也与PD相关,这与它们在神经炎症中的作用一致[24]。除黑质外,其他中脑和大脑区域一般也与PD有关,但确切的病因仍不清楚[25]。
smajiki等人对冷冻的人死后中脑(6名特发性PD患者和5名对照)进行了单核测序(snRNA-seq),以解码与特发性帕金森病相关的主要细胞类型:少突胶质细胞(ODC)、少突胶质细胞前体细胞(OPC)、小胶质细胞、星形胶质细胞、室管膜细胞、周细胞、内皮细胞、DaNs以及兴奋性、抑制性和gaba能神经元[15]。除了这11种细胞类型外,该研究还发现了一组额外的神经元细胞,它们几乎完全来自特发性PD中脑,其特征是编码钙依赖性分泌激活因子的CADPS2高表达(图3a)。这些细胞表现出与DaNs相似的表达谱,除了TH水平较低(TH是DaNs的标记基因,编码酪氨酸羟化酶,参与酪氨酸向多巴胺的转化)。这些cadps2高表达的细胞可能代表PD中退化的DaNs,但需要进一步的研究来证实这一假设。此外,本研究发现PD中ODC水平降低,星形胶质细胞和小胶质细胞增加。pd衍生的小胶质细胞的成像分析显示其分支减少和变形虫形状,表明其处于激活状态(图3a)。在特发性PD中,小胶质细胞表现出与炎症轨迹相关的基因(IL1B、GPNMB、HSP90AA1)的上调,这与它们在大脑中作为初级免疫细胞的作用一致。相比之下,ODC表现出应激诱导的S100B上调。
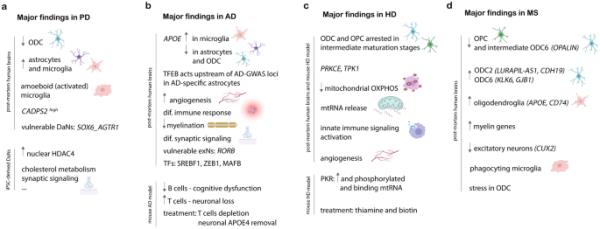
sc/snRNA-seq研究中最常见的神经退行性疾病的主要发现,包括帕金森病、阿尔茨海默病、亨廷顿病和多发性硬化症
另一项研究聚焦于DaNs,并开发了一种从人死后黑质中富集这种细胞类型的方案,使用荧光激活的细胞核分选富集nr4a2阳性细胞(该基因通过小鼠中脑scRNA-seq数据被鉴定为哺乳动物中脑神经元的标记)[26,27]。Kamath及其同事鉴定出10个DaN亚群,其中表达CALB1(6个亚群)和SOX6(4个亚群)作为一些主要的DaN标记基因[26]。为了研究DaNs的进化保守性,他们将人类数据集与其他物种(猕猴、北方树鼩、大鼠和小鼠)整合在一起,发现CALB1_GEM群体仅存在于人类和猕猴数据中,与这些物种具有更高的认知功能相一致。他们还发现,表达VIM_LHX2的星形胶质细胞亚群增加,对PD的退行性变化有反应,表达GPNMB的小胶质细胞亚群增加,这与smajiki等人的研究结果一致[15]。重要的是,通过snRNA-seq和单分子荧光原位杂交(smFISH)鉴定了表达SOX6_AGTR1的DaNs易感人群,这些细胞中富集了PD危险基因(SNCA、MAPT、GAK、WNT3、IGSF9B;图3 a)。一项对PD供者匹配的黑质和皮层样本的独立研究发现PD风险与DaNs和ODCs之间存在关联[28]。一般的DaN模块与线粒体过程、内吞作用、蛋白质泛素化和巨噬有关;ODC模块与基因调控、激酶活性、磷酸化、神经发生和代谢过程有关,而OPC风险相关模块则与基因调控、细胞分化和代谢过程有关。有趣的是,PD的遗传风险似乎不仅通过主要受影响的DaNs表现出来,还通过ODC和OPC表现出来,支持越来越多的证据表明胶质细胞在神经退行性疾病中的重要性。与此相一致的是,已知的PD基因LRRK2在OPC中的表达高于该数据集中其他神经细胞类型[28]。一项将全基因组关联研究(GWAS)结果与小鼠神经系统scRNA-seq数据相结合的研究进一步支持了这些观察结果,发现PD不仅与胆碱能神经元和单胺能神经元(多巴胺是一种单胺神经递质)相关,还与ODC和肠神经元相关[29]。肠道神经元与PD的相关性支持了Braak的假设,即特发性PD可能始于摄入的病原体,然后通过迷走神经从肠道扩散到大脑[30]。
另一种流行的研究PD分子病因的方法是使用ipsc衍生的dan[31]。Lang及其同事研究了来自葡萄糖脑苷酶基因GBA-N370S中PD风险变异个体的ipsc衍生的dan[32,33]。他们发现组蛋白去乙酰化酶4 (HDAC4)抑制许多基因(TSPAN7、ATP1A3、RTN1、PRKCB),导致内质网(ER)应激基因(PDI、FKBP9、ERO1A)上调[33]。有趣的是,虽然总HDAC4水平在PD中没有变化,但作者发现其在DaNs中特异性地增加了核定位,这与它在细胞核和细胞质之间移动的生理条件相反(图3a)。HDAC4定位或活性的药理学调节恢复了错误调控基因的表达水平,最终纠正了内质网应激表型以及自噬和溶酶体途径的扰动。这些关于GBA变异细胞系的发现可以扩展到其他特发性PD病例,这为PD提供了一种有希望的新治疗策略[33]。另一项研究调查了鱼藤酮(一种有毒的农药,通过诱导氧化应激引起PD样症状)、tunicamycin(引起内质网应激)和一种特定的SNCA变体对ipsc衍生的dan中PD表型发展的影响,并确定了成熟的dan易受细胞死亡的影响[34]。这些易感神经细胞具有多巴胺能神经元谱系基因(NR4A2、LMO3、POU2F2、LMX1A、DDC、DRD2)、PD基因(SNCA、MAPT、UCHL1、ATP13A2)、细胞骨架和神经元投射基因、微管蛋白基因等的表达特征。鱼烯酮处理后,该细胞群显示SNCA、氧化磷酸化(MT-CO1、MT-CO2、NDUFA5、NDUFB6)和胆固醇生物合成基因(SQLE、ACAT2、ACSL3、HMGCR)的表达降低。tunicamycin诱导内质网应激导致内质网应激反应基因(如预期)、翻译相关基因和热休克基因上调。另一方面,与胆固醇合成相关的基因被下调,与鱼藤酮治疗相似。非洛地平可以挽救氧化应激表型(由鱼tenone引起),但不能挽救内质网应激表型(由tunicamycin引起),此前有报道称非洛地平可以改善SNCA-A53T DaNs的表型[35]。使用基因组编辑产生的等基因SNCA-A53T细胞系,作者再次发现胆固醇代谢的扰动,以及突触信号、糖酵解和泛素蛋白体降解的变化。PD的主要研究结果总结于图3a。
阿尔茨海默病(AD)的典型特征是认知能力下降,是痴呆的最常见原因(尽管痴呆可以由其他神经退行性或心血管疾病引起)[36]。阿尔茨海默病影响的主要认知领域是记忆、语言、视觉空间和执行功能[2]。除了失忆的表现,年轻人也可能出现非失忆缺陷——被称为后皮层萎缩——如阅读、面部识别方面的挑战,或处理复杂视觉场景的困难[37]。AD的遗传危险因素包括APP(编码淀粉样蛋白前体蛋白)、PSEN1和PSEN2(分别编码早老素1和2)中罕见的显性变异,以及APOE中更常见但不完全渗透的变异,以及TREM2和MS4中的变异[2,38,39]。在病理学上,AD的特征是大脑皮层中存在含β-淀粉样蛋白斑块和含tau的神经原纤维缠结[40,41]。
One of the first and the largest studies that applied snRNA-seq on post-mortem AD brains profiled prefrontal cortex samples from 48 individuals with varying degrees of AD pathology and found that myelination-related processes were perturbed in multiple cell types [42]. The study by Mathys et al. revealed several unexpected findings including that large transcriptional changes occur early in the disease before the development of severe pathological features and that several AD-pathology-associated cell subpopulations were enriched in female cells. The authors also demonstrated that the AD risk factor APOE was upregulated in microglia, while it was downregulated in astrocytes (Fig. 3b). This finding emphasized the value of scRNA-seq methods as compared with bulk RNA sequencing, given that microglia were underrepresented in bulk data [42]. Some of these findings were replicated in an independent study, such as APOE upregulation in microglia and its downregulation in astrocytes and OPC, and increased LINGO1 levels in AD-specific subclusters [43]. Furthermore, Grubman and colleagues found that the transcription factor EB (TFEB), a regulator of lysosomal function and autophagy, acts upstream of 10 GWAS loci for AD (BIN1, CLDN11, POLN, STK32B, EDIL3, AKAP12, HECW1, WDR5, LEMD2, DLC1) in a disease-specific astrocyte subpopulation (Fig. 3b). Another study reported the relevance of endothelial cells in AD, in addition to the previously mentioned cell types [44]. Lau et al. observed an enhanced angiogenesis in endothelial cells, together with aberrant immune response in endothelial cells and microglia, reduced myelination in ODC, and impaired synaptic signaling in neurons and astrocytes (Fig. 3b) [44]. Three AD-upregulated subpopulations expressed genes associated with angiogenesis (CLDN5, ERG, FLT1, VWF) and antigen presentation (MHC-I complex). Analysis of bulk microarray data from a mouse AD model revealed a similar transcriptomic profile, suggesting that the activation of endothelial cells in neurodegeneration is conserved between humans and mice [44]. A study on 10 male individuals with APOE ε3/ε3 genotype that sequenced a large amount of nuclei (10,000 per individual) in entorhinal cortex (affected early in the disease progression) and superior frontal gyrus (affected late in the disease progression) identified RORB as a marker of selectively vulnerable excitatory neurons in the entorhinal cortex (Fig. 3b) [45]. The same study found an increased amount of microglia with AD progression (microgliosis) and reactive astrocytes that showed downregulation of genes associated with homeostasis. A simultaneous profiling of chromatin accessibility and gene expression on post-mortem prefrontal cortex samples identified cell-type-specific cis and trans regulatory elements and their target genes in AD [46]. Morabito and colleagues examined the regulatory roles of transcription factors SPI1 in microglia and NRF1 in ODC and found that SPI1 acts as a transcriptional repressor in late-stage AD, while NRF1 may contribute to neuronal dysfunction through the disruption of myelination. SREBF1 that regulates cholesterol and fatty acid metabolism was identified as a transcriptional activator throughout the ODC trajectory, while the APOE locus had cis-regulatory chromatin networks altered in AD in microglia and astrocytes [46]. Additional transcription factors that might play a role in AD-specific gene regulation include ZEB1 in neurons and MAFB in microglia (Fig. 3b) [47]. A very recent study on parietal cortex samples from AD autosomal dominant (APP and PSEN1) and risk-modifying variant (APOE, TREM2, MS4A) carriers detected the affected pathways: APOEε4 inhibitory neurons displayed signs of ferroptosis (an iron-dependent form of cell death), TREM2 ODC showed a dysregulated autophagy-lysosomal pathway, while MS4A microglia had dysregulated genes of the complement cascade [48]. The relevance of the human brain vasculature in AD has also been demonstrated in a study that found selective vulnerability of extracellular matrix-maintaining pericytes and gene expression patterns that implicated dysregulated blood flow [49]. Moreover, many of the AD GWAS genes were found to be expressed in the brain vasculature, and they were associated with endothelial protein transport, adaptive immune system, and extracellular matrix pathways [49]. scRNA-seq of peripheral blood mononuclear cells (PBMC) reported a decrease in B cells in individuals with AD, where the reduction in B cells correlated with the patients’ clinical dementia rating scores. These results were confirmed in a mouse AD model, where the B cell depletion accelerated cognitive dysfunction and worsened the phenotype [50]. Mice with tauopathy (but not with amyloid beta deposition) developed a unique innate and adaptive immune response, where numbers of T cells were increased in areas with tau pathology and could be correlated with the extent of neuronal loss. Depletion of T cells by peritoneal administration of neutralizing antibodies led to strong depletion of CD4+ and CD8+ T cells in brain parenchyma, meninges, and peripheral blood, blocking tau-mediated neurodegeneration. Furthermore, microglia shifted from activated toward homeostatic state after T cell depletion [51]. Another successful approach in treating AD-specific cellular phenotypes in mice with tauopathy was a selective removal of neuronal APOE4, which led to a reduction in tau pathology, gliosis, neurodegeneration, neuronal hyperexcitability, and myelin deficits [52]. All major findings on AD are summarized in Fig. 3b.
亨廷顿舞蹈病(HD)的特征是一种称为舞蹈病的多动运动障碍,并伴有痴呆、行为和精神障碍[4]。它是由亨廷顿蛋白基因(HTT)外显子1的显性遗传CAG重复扩增引起的。纹状体的中棘神经元是最易受突变HTT影响的细胞群,尽管大脑皮层也会发生神经元功能障碍和死亡。对人类死后和小鼠HD样本纹状体和皮层的snRNA测序发现,少突胶质细胞谱系(odc和opc)在中间成熟阶段被阻滞[53]。Lim及其同事发现PRKCE(编码蛋白激酶C epsilon)和TPK1(编码硫胺素焦磷酸激酶1)是这些过程中的中心基因(图3c)。在HD小鼠模型中,大剂量硫胺素和生物素治疗可挽救神经元转录失调,改变ODC和OPC发育基因[53]。中棘神经元对突变HTT的敏感性增加也可以通过下调线粒体氧化磷酸化途径、释放线粒体RNA和激活先天免疫信号来解释[54]。在小鼠HD模型纹状体中,先天免疫传感器蛋白激酶R (PKR)上调和磷酸化,表明其激活(图3c)。此外,与先前的研究结果一致[55],线粒体RNA可以通过免疫沉淀直接从PKR复合物中纯化出来。一项针对星形胶质细胞的研究发现,它们在HD中表现出可变的转录表型:其中一些细胞的金属硫蛋白和热休克基因上调,而另一些细胞的胶质纤维酸性蛋白(一种反应性星形胶质细胞标志物)上调。另一方面,一些正常的原生质星形胶质细胞基因和脂质合成基因的表达减少[56]。一项关注人类脑血管重要性的研究发现,星形胶质细胞亚群和小胶质细胞亚群中高表达的基因与血管生成和血管内皮细胞迁移的调节有关,这表明这些胶质细胞可能与血管调节有关[57]。本研究还发现内皮细胞(IKBKB、IRF2/3、STAT3)、星形胶质细胞和小胶质细胞中一些先天免疫激活基因上调。另一方面,他们观察到血脑屏障(BBB)紧密连接蛋白(CLDN5和TJP1)的减少,这是已知导致血脑屏障完整性丧失的原因。一项HD小鼠模型纹状体的snRNA-seq研究检测了易感细胞类型(中棘神经元、ODC和星形胶质细胞)中基因的异常表达,发现许多细胞类型识别基因在其原代细胞类型中下调,而在其他纹状体细胞类型中异常上调[58]。HD的主要研究结果总结于图3c。
多发性硬化症(MS)是一种炎症性、脱髓鞘性神经退行性疾病,受遗传和环境因素的影响。病理特征为在脑和脊髓内形成脱髓鞘病变[5]。在这种疾病中受影响的主要细胞类型是少突胶质细胞,因为它们提供代谢支持和轴突髓鞘。与前面提到的常见ND相比,这种疾病的研究较少,特别是使用单细胞测序方法。一项对死后人类大脑白质区域进行snrna测序的研究发现,MS病变中OPC(表达PDGFRA、BCAN和SOX6)和中间ODC亚群(表达OPALIN和LINC00844)的核数量减少。另一方面,其他ODC集群(集群2表达LURAPIL-AS1和CDH19;表达KLK6, GJB1)和少突胶质细胞(表达APOE和CD74)的簇5在MS中富集,有趣的是,在MS成熟的ODC中,一些髓鞘蛋白基因被上调,这表明这些细胞的一个子集有助于髓鞘再生[59]。另一项研究发现,在皮质脱髓鞘的MS样本中,cux2表达的兴奋性神经元数量减少,并存在选择性易感性。易感兴奋性神经元中上调的基因包括氧化应激、线粒体功能障碍、细胞死亡途径基因以及长链非编码rna (BCYRN1和LINC00657)。ms -髓鞘化ODC中上调的基因与热休克反应、细胞应激、铁积累、泛素介导的蛋白质降解等相关,表明ms中ODC存在严重的应激。此外,该研究还发现了一群富含ODC特异性标志物的吞噬小胶质细胞[60]。图3总结了MS的主要发现。
特定细胞(亚)群体的选择性易感性是神经退行性疾病的一个标志,但迄今为止,技术缺乏必要的吞吐量和分辨率来大规模研究这些变化[61]。Sc/ snrna组学方法为研究这些细胞类型特异性变化提供了强有力的方法[62,63,64]。近年来高通量单细胞测序工作流程和计算工具的发展使该技术在生命科学的许多领域得到应用,加上测序成本的降低,为其广泛应用提供了巨大的希望。该技术正处于人类遗传学和病理学诊断的门槛,有可能将其整合到常规临床诊断和个性化医疗中[16,17]。然而,它在神经退行性疾病方面的应用仍然存在一些挑战:首先是组织获取,因为脑组织取样的可及性较低。其次,sc-RNA测序在福尔马林固定和石蜡包埋的组织中仍然很困难,就像死后样本一样[65,66]。这些问题至少可以通过使用患者来源的iPSC和类器官来部分克服,特别是在分化方案正在开发和不断改进的情况下[67]。第三,sc-seq的一个相当大的缺点是在分离单个细胞/细胞核时丢失拓扑信息,但最近出现的空间转录组学方法可以添加这层缺失的组织复杂性信息。最后,尽管多项研究已经对相同的组织进行了测序,但缺乏单个细胞(亚)类型的通用命名法是未来需要解决的一个挑战[68]。
通过联合sc-模式(例如转座酶可及染色质(ATAC)和RNA, DNA甲基化和ATAC,空间转录组学,sc-蛋白质组学,sc-代谢组学等)和更大的样本量,sc-seq组学方法将进一步扩展我们对细胞类型和整个器官如何影响健康和疾病的认识。重要的是,引入全脑甚至整个生物体单细胞图谱,每个人都可以浏览(即使没有先前的生物信息学知识),这是向前迈出的重要一步[12,69,70]。总之,这些努力将使人们能够更好地了解一种疾病的潜在机制,并使最终能够针对受影响人群和途径进行治疗。一旦已知受影响的细胞类型及其特定的基因/蛋白质,就可以通过改变基因表达(例如,通过使用病毒和非病毒载体或反义寡核苷酸)或蛋白质水平(例如,通过使用目标特异性抗体)来调节它们的水平,或通过再生医学手段替换受损细胞。尽管考虑到脑组织的可及性和血脑屏障的渗透性,这些方法对ND仍然具有挑战性,但随着科学知识的扩展,治疗选择将变得可行。


发表评论
暂时没有评论,来抢沙发吧~